Submitting Institution
Queen Mary, University of LondonUnit of Assessment
Clinical MedicineSummary Impact Type
HealthResearch Subject Area(s)
Biological Sciences: Genetics
Medical and Health Sciences: Neurosciences
Summary of the impact
Research at the Centre for Cutaneous Research at Queen Mary has led to
gene discovery and
molecular diagnosis for a number of single gene skin disorders and
associated syndromes
including hearing loss, inflammatory bowel disease, cardiomyopathy and
oesophageal cancer. It
has identified GJB2 mutations (encoding Cx26) as major cause of
genetic hearing loss (20-50% of
all cases) and ABCA12 mutations with the (often fatal) recessive
skin condition Harlequin
Ichthyosis. Impacts include: 1) increased medical and scientific
awareness/knowledge of the
inherited basis of these conditions, 2) changes in clinical practice and
molecular diagnosis, 3)
improved information for patients, parents and the public.
Underpinning research
Basic genetic research in the Centre for Cutaneous Research at Queen Mary
includes:
GJB2 and hearing loss (discovery 1996-1997). In 1997, the
team hypothesised that hearing loss
in families with autosomal dominant skin disease may be due to an
unidentified gene mapping in a
locus for recessive non-syndromic hearing loss on chromosome 13 (termed
DFNB1). Kelsell et al
sequenced GJB2 encoding Connexin26 and identified mutations linked
to hearing loss in this
family. They then sequenced recessive non-syndromic hearing loss families
and identified further
GJB2 mutations. This group (including Professor Irene Leigh) were
the first to link GJB2 (encoding
Cx26) mutations with hearing loss. The paper, published in Nature
in 1997 and cited by over 1,000
publications since, described the first autosomal non-syndromic deafness
gene ever identified [1].
The implication of this finding is that GJB2 mutations account for
between 20 - 50% of all genetic
hearing loss in many distinct geographic and ethnic populations. Cochlear
implants in children with
GJB2 hearing loss lead to major clinical improvement in hearing
status, particularly if done in the
early years of life (eg PMID: 11977173).
The linking of GJB2 mutation and hearing loss led to further
studies by the Kelsell group including
GJB2 mutation screening, the development of molecular assays for
rapid mutation screening and
tissue- / cell-based functional assays for specific mutations. Dominant
mutations in GJB2 and other
connexin encoding genes including Cx31 were also shown to underlie
cutaneous conditions with
and without hearing loss [2]. In addition, the team provided evidence that
the high carrier frequency
of recessive GJB2 mutation may be linked to heterozygous
advantage, conferring protection from
pathogen-borne disease. Functional studies are revealing new roles for
loss of functional Cx26
leading to improved epithelial barriers and impaired bacterial entry into
epithelial cell, thus another
potential area of medical benefit.
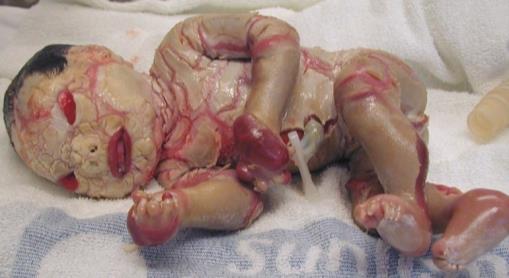
") Harlequin Ichthyosis and ABCA12 (discovery 2005-2006).
Harlequin Ichthyosis and ABCA12 (discovery 2005-2006).
Harlequin Ichthyosis (HI) is the most severe form of congenital
ichthyosis. Appearance at birth is
usually characteristic (left panel above). The whole body is encased in an
armour of thick white
plates of scale separated by deep red fissures. The upper and lower
eyelids are usually retracted,
causing bilateral ectropion; the lips are parted, causing eclabium. The
nose is flattened and
appears rudimentary. Babies who survive infancy and into adulthood develop
skin changes
resembling a severe congenital ichthyosiform erythroderma (right panel).
By 2005, the Kelsell Group at Queen Mary (including Professor Edel
O'Toole) had identified and
recruited a number of families worldwide with apparent recessive HI. Using
the facilities at Barts
and The London Genome Centre, they implemented a high density Single
Nucleotide
Polymorphism (SNP) array / homozygosity approach to map the condition to a
region on
chromosome 2q35 [3]. Positional candidate gene sequence analysis led them
to be the first group
to identify ABCA12 mutations as the cause of this disease. They and others
screened HI families
with identifiable biallelic ABCA12 mutations, which account for over 98%
of cases. Kelsell's centre
is now a global referral centre for the genetic diagnosis of HI; they have
now identified ABCA12
mutations in over 130 families [4-6]. Furthermore, a functional programme
of work has been
established with the development of HI keratinocyte cell lines and in
vitro 3D human HI skin
models to dissect the cellular pathways disrupted by loss of ABCA12 and
the development of
assays which may lead to new therapies.
Funding: This research has been supported from multiple sources,
including BBSRC, Barts and
the London Charity, Wellcome Trust, Action Medical Research, British Skin
Foundation, Ichthyosis
Support Group, EU Network consortium (Nanodrug) and SHhIRT (Samuel
Hardgrave Harlequin
Ichthyosis Research Trust).
References to the research
1. Kelsell DP, Dunlop J, Stevens HP et al. Connexin 26 mutations
in hereditary non-syndromic
sensorineural deafness. Nature 1997; 387, 80-83. PMID: 9139825
2. Scott CA, Kelsell DP. Key functions for gap junctions in skin
and hearing. Biochemical Journal
2011; 438: 245-54. PMID: 21834795.
3. Kelsell DP, Norgett EE, Unsworth H et al. Mutations in ABCA12
underlie the severe congenital
skin disease Harlequin Ichthyosis. American Journal of Human Genetics
2005; 76: 794-803.
PMID: 15756637.
4. Thomas AC, et al and Kelsell DP. Novel and recurring ABCA12
mutations associated with
Harlequin Ichthyosis: implications for prenatal diagnosis. British
Journal of Dermatology 2008;
158: 611-3. PMID: 17986308.
5. Scott CA et al and Kelsell DP. Targeted sequence capture and
high-throughput sequencing in
the molecular diagnosis of ichthyosis and other skin diseases. Journal
of Investigative
Dermatology 2012; 133: 573-6. PMID: 22992804.
6. Rajpopat S, et al and Kelsell DP, O'Toole EA. Harlequin
ichthyosis: a review of clinical and
molecular findings in 45 cases. Archives of Dermatology 2011; 147:
681-6. PMID: 21339420.
Details of the impact
Improved clinical practice, diagnostic accuracy and public awareness
in relation to GJB2
and hearing loss
Since GJB2 accounts for up to 50% of genetic hearing loss, most
clinical genetic services and
commercial testing companies now offer GJB2 testing, including
23andMe, University of Chicago
Genetic Services, University of Iowa, ARUP Laboratories, Greenwood Genetic
Centre, Knight
Diagnostic Centre, Harvard Medical School, Athean Diagnostics and
Centogene. In addition,
bloodspot-based genetic testing for GJB2 alleles can provide a
means for rapid confirmation in the
subset of infants who fail bedside newborn hearing tests [7]. In a US
study in 2000, eight
participating laboratories provided GJB2 testing to approximately
230 families per month [8]. These
studies have led to a number of additional impacts:
- Improved medical and scientific awareness and knowledge of GJB2
mutations as a major
underlying cause of congenital hearing loss [9].
- Changes in clinical practice and molecular diagnosis:
a. Early diagnosis allows for early intervention such as cochlear
implants in patients with
GJB2 associated hearing loss. In the UK for example, 1,650 GJB2
tests were performed in
2010-2011 according to the Clinical Molecular Genetics Society Audit
[10]. Delayed
diagnosis of hearing loss in infants may have a harmful effect on
social, emotional,
cognitive, and academic development. A genetic diagnosis provides key
information for
genetic counseling, including prognosis and recurrence risk. For
example, not all GJB2-associated
hearing loss presents at birth [11].
b. A positive diagnosis helps rule out other diseases, including Ushers
Syndrome.
- Genetic testing as described above is now recommended as the gold
standard by the
European Molecular Genetics Quality Network, and laboratories that
follow this guidance can
seek a formal certificate of quality [12].
- Improved clinical and genetics information on GJB2-associated
hearing loss for patients with
hearing loss and their families via websites, leaflets and similar
material [13].
Improved diagnosis, management, prenatal diagnosis and awareness of
Harlequin
ichthyosis
Kelsell et al's 2005 discovery of recessive ABCA12 mutations as
the major cause of this
devastating and often life threatening skin condition has led to a number
of impacts:
- Molecular diagnosis. The team has developed into a major global
referral centre for HI in the
eight-year period since they discovered the HI gene. Over 130 families
from the UK, Europe,
Africa and Asia have been sent for ABCA12 genetic analysis to this
laboratory for molecular
diagnosis. In 98% of these cases, ABCA12 mutations were identified. For
many of the samples
from overseas there is often no financial support for the HI families to
cover the genetic test
and in these cases Kelsell's team do the tests at no cost to the family.
Importantly, they have
developed genetic screening strategies such as copy number arrays,
targeted and exome high
throughput sequencing to increase accuracy of mutation detection and
increase turnaround
time (within three months), which is critical for many HI families. Last
year, they performed
genetic analysis on eleven HI families, mainly from Pakistan, India and
Iran. For UK, Europe
and the US, the ABCA12 genetic test is now an established service in
general clinical genetic /
skin disease laboratories.
- Pre-natal diagnosis (in excess of 20 HI families by this laboratory)
based on the genetic
findings. As predicted, in around 75% of cases, the test indicated that
the mother was carrying
a child that would not develop HI, and this was of great reassurance to
the family. Furthermore,
the team has performed what is believed to be the first case of
pre-implantation genetic
diagnosis for this condition, allowing the single genetically unaffected
embryo to be selected
from nine fertilised eggs, hence producing a child who was not only
phenotypically normal but
who was not even a carrier of the condition. This clinical application was
published as an online
communication by Barts Charity [14].
- Data to support specific changes to clinical management. As the team
had access to a large
cohort of HI families (due to families requesting genetic testing), the
team were able to perform
the first large-scale study of HI, providing important prognostic
information for clinicians and
affected families. Previously the only clinical information were
individual case reports
describing neonatal lethality. The large number of HI families in this
study published in the
Archives of Dermatology (reference 6 above) provided huge insights into
biology of this severe
skin disease including that nearly 50% of HI babies in our study survived
(eg retinoids give
86% survival).
- Patient and public awareness. Kelsell et al's work on HI has
been a component in three TV
documentaries (ITV, Channel 5 and Discovery) shown worldwide [15] and also
as an
interactive learning tool within Centre of the Cell, QMUL's interactive
science education centre
[16].
Sources to corroborate the impact
GJB2 and hearing loss
- Schimmenti LA, Warman B, Schleiss MR et al. Evaluation of
newborn screening bloodspot-based
genetic testing as second tier screen for bedside newborn hearing
screening. Genetic
Medicine 2011; 13:1006-10.
- Kenneson A, Myers MF, Lubin IM, Boyle C. Genetic laboratory practices
related to testing of
the GJB2 (Connexin-26) gene in the United States in 1999 and 2000. Genetic
Testing 2003; 7:
49-56.
- Ardle BM, Bitner-Glindzicz M. Investigation of the child with
permanent hearing impairment.
Archives of Diseases of Childhood (Education and Practice Edition)
2010; 95: 14-23.
- Clinical Molecular Genetics Society Audit 2010-2011:
http://www.cmgs.org/CMGS%20audit/2011%20audit/CMGSAudit10_11_FINAL.pdf
- Minami SB, Mutai H, Nakano A, Armoto Y et al. GJB2-associated hearing
loss undetected by
hearing screening of newborns. Gene 2013 Sep 5, epub ahead of
print. doi:
10.1016/j.gene.2013.08.094.
- Guidelines for testing of DFNB1 as part of EMQN. Hoefsloot LH, Roux
A-F and Bitner-Glindzicz
M on behalf of the contributors to the EMQN DFNB1 best practice meeting.
European Journal
of Human Genetics advance online publication, 22nd May
2013. doi: 10.1038/ejhg.2013.83.
- Example of patient support organisation website for GJB2-related
deafness.
https://www.counsyl.com/diseases/gjb2-related-dfnb-1-nonsyndromic-hearing-loss-and-deafness/
ABCA12 and Harlequin Ichthyosis
- First recorded case of pre-implantation genetic diagnosis of HI,
leading to confirmation that
fetus was unaffected: www.bartsandthelondoncharity.org.uk/News/Detail/Look-what-we-got-when-we-googled-genetics
- TV Documentaries, featuring Kelsell et al's research. For
example: ITV1 `Real Lives' October
2005; `The Girls with Too Much Skin' www.channel5.com/shows/extraordinary-
people/episodes/extraordinary-people-the. This programme was
short-listed for the Media
Award for factual Programming at RADAR's People of the Year Human Rights
Awards 2008.
Another documentary of this research including gene identification and
skin models of
Harlequin Ichthyosis was filmed October 2012 and will be shown in late
2013 on the Discovery
Channel.
Other media coverage: BBC Online http://news.bbc.co.uk/1/hi/health/4337009.stm
- Public understanding of science. The School of Medicine and
Dentistry's Centre of the Cell
`Find a Gene' interactive exhibit featuring HI: www.centreofthecell.org/centre/?page_id=129